# QTL 数量性状位点 (QTL) 是与特定表型性状相关的 DNA 区域,其程度各不相同,可归因于多基因效应,即两个或多个基因的产物及其环境。这些QTL通常位于不同的染色体上。解释表型特征变异的QTL数量表明了一个特征的遗传结构。这可能表明植物高度受到许多效果较小的基因控制,或者受到少数效果较大的基因控制。 通常,QTL是连续性状(那些连续变化的性状,例如身高)的基础,而不是离散性状(具有两个或几个性状值的性状,例如人类的红头发、隐性性状或孟德尔在他的实验中使用的光滑与起皱的豌豆)。 而且,单个表型性状通常由许多基因决定。因此,许多 QTL 与单个性状相关。QTL 的另一个用途是鉴定一个性状的候选基因。然后可以将该区域任何基因的 DNA 序列与功能已知的基因的 DNA 数据库进行比较,这一任务是标记辅助作物改良的基础。 # eQTL 表达数量性状位点(expression quantitative trait locus, eQTL)是一类能够影响基因表达量的遗传位点(大部分都是单核苷酸多态性,SNP),具有一定的生物学意义。迄今为止最全的eQTL数据库是**GTEx([https://www.gtexportal.org/home/](https://link.zhihu.com/?target=https%3A//www.gtexportal.org/home/))**,如今已更新到第八版了。 一般而言,eQTL主要分为两类:(1)顺式eQTL(cis-eQTL):它主要是指与所调控基因相距较近的eQTL,一般多位于所调控基因的上下游1Mb区域;(2)反式eQTL(trans-eQTL):与cis-eQTL恰恰相反,反式是指距离所调控基因位置比较远的eQTL,有时候距离甚至超过5Mb。因此,对于eQTL分析而言,我们通常需要考虑两点,**SNP和基因表达水平的关联度以及SNP与基因的距离**。 由于大量eQTL数据库的开发,我们现在可以直接利用别人的结果寻找SNP调控的基因,最常用的就是GTEx数据库了,大家可以自行学习了解。接下来我将介绍如何利用自己的数据计算并确定相关eQTL。 利用原始数据做eQTL分析,我们至少需要三个文件,第一个是**样本信息文件**,该文件包含样本的年龄,性别和人种等等;第二个是**基因表达量文件**,它表示的是每个基因在每个样本中的表达含量;第三个是**基因型数据**,也即每个样本的基因型数据。 接下来我们看看数据格式: 第一个是样本信息文件,除开第一列,其它列都代表不同的样本,每一行代表的是样本的表型信息。  第二个是基因表达量信息,同样地,除开第一列,其它列都代表不同的样本,每一行代表的是不同的基因(一般来说基因表达数据需要先进行标准化转换)。 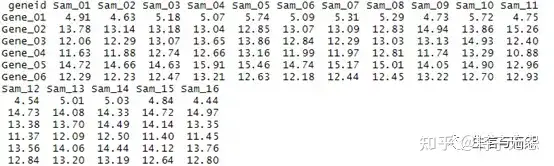 第三个是基因型数据,同样地,除开第一列,其它列都代表不同的样本,每一行代表的是不同的基因型(SNP),一般基因型数据用0,1,2这三个数字编码,代表的是效应等位基因剂量。举个简单的例子,SNP1的等位基因分别是A和C,如果我们以A为效应等位基因,那么基因型AA的剂量便是2,AC为1,CC为0。 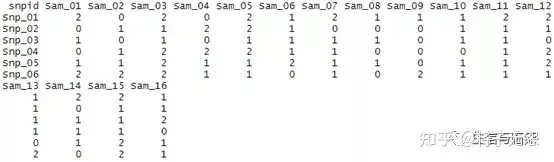 有了这些数据,我们就可以简单分析SNP和基因表达量的关系了 其数学模型如下: gene1 ~ snp1 + sex + age + error_term 这里gene1(因变量)一般就是一个基因的表达量,snp1(自变量)就是一个SNP的基因型,两者拟合,矫正相关干扰项(如sex和age等),error_term是指回归模型的误差项。 如果想区分顺式还是反式eQTL,这时候就需要结合基因与SNP的位置信息了。 Loading... # QTL 数量性状位点 (QTL) 是与特定表型性状相关的 DNA 区域,其程度各不相同,可归因于多基因效应,即两个或多个基因的产物及其环境。这些QTL通常位于不同的染色体上。解释表型特征变异的QTL数量表明了一个特征的遗传结构。这可能表明植物高度受到许多效果较小的基因控制,或者受到少数效果较大的基因控制。 通常,QTL是连续性状(那些连续变化的性状,例如身高)的基础,而不是离散性状(具有两个或几个性状值的性状,例如人类的红头发、隐性性状或孟德尔在他的实验中使用的光滑与起皱的豌豆)。 而且,单个表型性状通常由许多基因决定。因此,许多 QTL 与单个性状相关。QTL 的另一个用途是鉴定一个性状的候选基因。然后可以将该区域任何基因的 DNA 序列与功能已知的基因的 DNA 数据库进行比较,这一任务是标记辅助作物改良的基础。 # eQTL 表达数量性状位点(expression quantitative trait locus, eQTL)是一类能够影响基因表达量的遗传位点(大部分都是单核苷酸多态性,SNP),具有一定的生物学意义。迄今为止最全的eQTL数据库是**GTEx([https://www.gtexportal.org/home/](https://link.zhihu.com/?target=https%3A//www.gtexportal.org/home/))**,如今已更新到第八版了。 一般而言,eQTL主要分为两类:(1)顺式eQTL(cis-eQTL):它主要是指与所调控基因相距较近的eQTL,一般多位于所调控基因的上下游1Mb区域;(2)反式eQTL(trans-eQTL):与cis-eQTL恰恰相反,反式是指距离所调控基因位置比较远的eQTL,有时候距离甚至超过5Mb。因此,对于eQTL分析而言,我们通常需要考虑两点,**SNP和基因表达水平的关联度以及SNP与基因的距离**。 由于大量eQTL数据库的开发,我们现在可以直接利用别人的结果寻找SNP调控的基因,最常用的就是GTEx数据库了,大家可以自行学习了解。接下来我将介绍如何利用自己的数据计算并确定相关eQTL。 利用原始数据做eQTL分析,我们至少需要三个文件,第一个是**样本信息文件**,该文件包含样本的年龄,性别和人种等等;第二个是**基因表达量文件**,它表示的是每个基因在每个样本中的表达含量;第三个是**基因型数据**,也即每个样本的基因型数据。 接下来我们看看数据格式: 第一个是样本信息文件,除开第一列,其它列都代表不同的样本,每一行代表的是样本的表型信息。  第二个是基因表达量信息,同样地,除开第一列,其它列都代表不同的样本,每一行代表的是不同的基因(一般来说基因表达数据需要先进行标准化转换)。  第三个是基因型数据,同样地,除开第一列,其它列都代表不同的样本,每一行代表的是不同的基因型(SNP),一般基因型数据用0,1,2这三个数字编码,代表的是效应等位基因剂量。举个简单的例子,SNP1的等位基因分别是A和C,如果我们以A为效应等位基因,那么基因型AA的剂量便是2,AC为1,CC为0。  有了这些数据,我们就可以简单分析SNP和基因表达量的关系了 其数学模型如下: gene1 ~ snp1 + sex + age + error_term 这里gene1(因变量)一般就是一个基因的表达量,snp1(自变量)就是一个SNP的基因型,两者拟合,矫正相关干扰项(如sex和age等),error_term是指回归模型的误差项。 如果想区分顺式还是反式eQTL,这时候就需要结合基因与SNP的位置信息了。 最后修改:2024 年 02 月 04 日 © 允许规范转载 赞 如果觉得我的文章对你有用,请随意赞赏