# 文献汇报 ## Title A reference map of potential determinants for the human serum metabolome 人血清代谢组潜在决定因素指南 https://doi.org/10.1038/s41586-020-2896-2 ## Abstract 1. 血清代谢组包含大量内源或外源的生物标志物或者疾病致病因子 1. 通常,化合物来源已知 1. 遗传性,基因,血液 2. 肠道微生物 3. 生活方式,吸烟或饮食 2. 大多数代谢物的key determinants知之甚少 2. 测量491个独特和deeply phenotyped的健康人血清样本中的1251个代谢物水平 3. ML用于预测个体的代谢物水平 1. 数据来源:遗传信息、肠道菌群、临床、饮食、生活方式和人体测量数据 2. 预测结果:超过76%的差异代谢物 3. 预测效力:饮食和肠道微生物组的预测能力最强 4. 总结:超过800代谢物的变化具有实际的意义,为理解不同条件下代谢物的变化机制和设计控制循环代谢物水平的干预措施铺平了道路 > 血清代谢组的主要研究对象是血清所含有的各种生物标志物。这些标志物既可是内源生成,也可由环境中摄取,其中部分标志物的来源较为清晰,包括高遗传性的代谢物或可被肠道菌或生活方式 (如吸烟或饮食) 所影响的代谢物,因此,血清代谢组研究对于了解多种疾病具有重要意义。 1. 首先介绍一下作者,Eran Segal,计算生物学领域的一个牛人,95-98年特拉维夫大学的理学学士学位,99-04年,斯坦福大学计算机科学和遗传学博士学位,导师Daphne Kolle,毕业后于洛克菲勒大学就职博后。博士毕业后的四年时间里,已经作为一作和通讯发表了三篇nature正刊和十几篇子刊。至今,共计近30篇NCS,子刊无数。最开始主要做转录相关方面的工作,08年左右开始做表观遗传的相关工作,随后开始进入微生物组和代谢组学的多组学结合领域。 2. 然后介绍一下文章的相关背景。血清代谢组的主要研究对象是血清所含有的各种生物标志物。这些标志物既可是内源生成,也可由环境中摄取,其中部分标志物的来源较为清晰,包括高遗传性的代谢物或可被肠道菌或生活方式 (如吸烟或饮食) 所影响的代谢物,因此,血清代谢组研究对于了解多种疾病具有重要意义。 3. 为更进一步了解血清代谢物的关键决定因素,2020年11月11日,来自以色列Weizmann科学院Eran Segal团队的研究人员在Nature杂志在线发表了题为A reference map of potential determinants for the human serum metabolome的研究论文,使用质谱技术对健康志愿者的血清样品进行了深度鉴定,基于志愿者的宿主遗传学、肠道微生物、临床参数及生活方式等特征,使用机器学习算法对血清样品中一千多种独特代谢物进行预测,从而揭示了主要代谢物的关键决定因素,从而更好地帮助我们了解这些代谢物在不同条件下变化机制,从而制定干预措施。 4. 我们看到方法概览 1.样本来源:491个健康的以色列人,18~70岁之间。 2. 数据采集:粪便、血清的宏基因组和代谢组、饮食、临床、生活方式 3.分析方式:非靶液质联用分析1251种代谢物,包括脂类、氨基酸、外源化合物、碳水化合物、肽和核苷酸,以及30%的未知化合物 ;设计预测代谢物通路的模型以对未知代谢物分类。 4.预测结果验证:对比测量数据和预测的数据,一致性高,重复性好;不同人的血清样本,并无相关性。 5.总结:本研究获得的代谢谱,提供了一种可揭示循环血代谢物的潜在决定因素的预测模型,帮助我们了解健康和疾病状态下的分子机制,并帮助我们寻找改变这些血清代谢物水平的新干预措施。 接着,分析结果 ## Main ### 方法概览 1. 临床、饮食、生活方式、遗传和肠道菌群数据事先收集 2. 非靶分析1251个代谢物水平,包括脂类、氨基酸、外源化合物、碳水化合、肽和核苷酸,以及30%的未知化合物 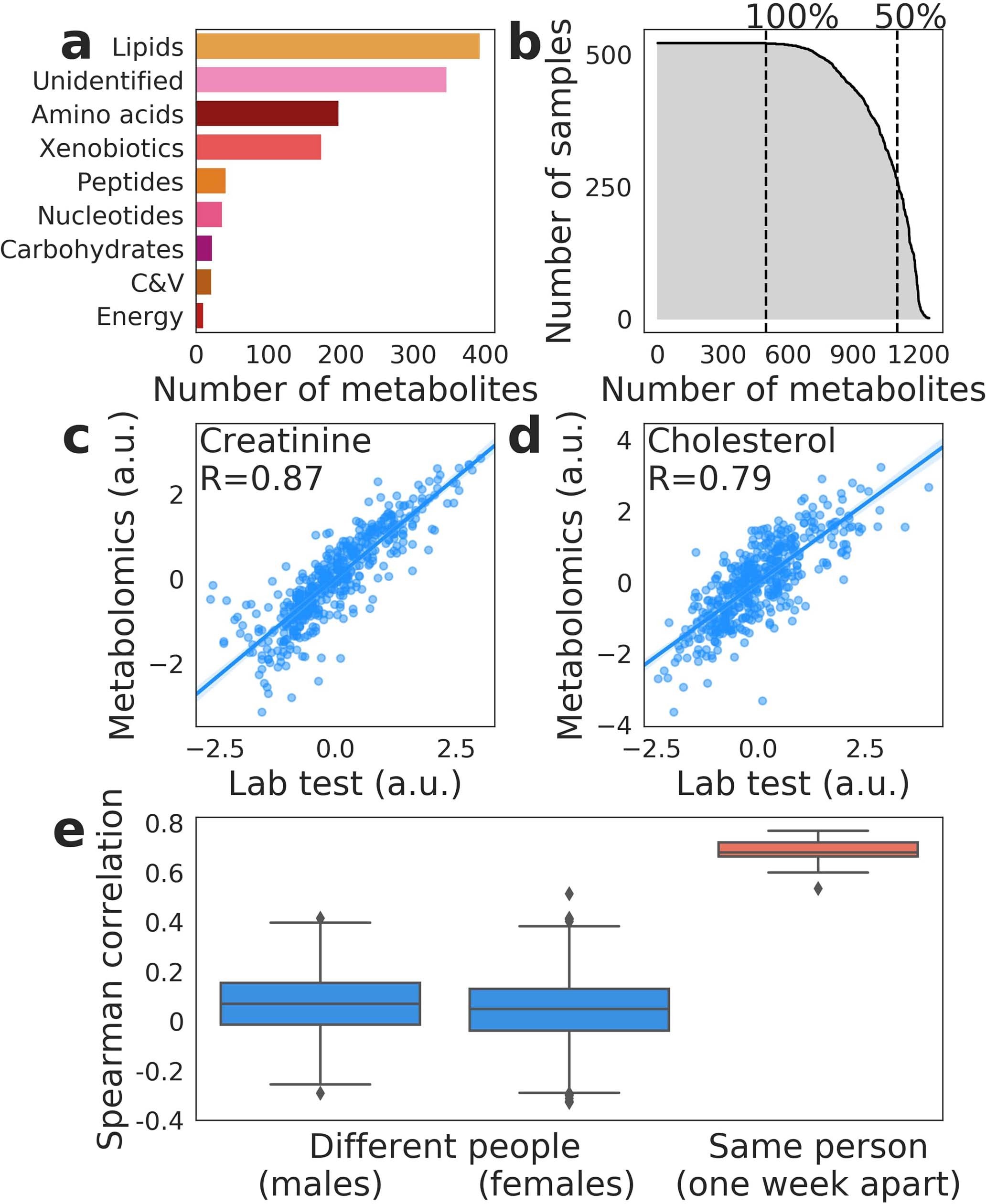 3. 设计可准确预测代谢物候选生物途径的模型 1. 所有样本均检测到498个代谢物,超过50%的样本中检测到1104个代谢物 2. 筛选出475名高质量数据 ## 血清代谢物的预测 > 可解释方差:很少有资料提到,以下是从代码中解读的公式,可能和实际的有些许差别。 > 可解释方差指标衡量的是所有预测值和样本之间的差的分散程度与样本本身的分散程度的相近程度。本身是分散程度的对比。最后用1-这个值,最终值越大表示预测和样本值的分散分布程度越相近。 > 1- 样本值与预测值之差的方差/样本方差 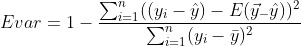 1. 血清代谢物的预测 1. 预测方法:gradient-boosted decision trees[^1],基于可释方差的系统线性模型预测志愿者数据 2. 预测结果:335个代谢物个别饮食相关特征所解释,182个代谢物可被肠道菌相关特征解释,因此**该模型对血清代谢物具有较强预测性**,Fig 1 ab。 2. 随后检测代谢物是否富集于主要预测因素superior prediction 1. 结果,Fig 1 c: 1. 临床数据能更好地预测血脂、氨基酸及多肽等类别的代谢物 2. 肠道菌数据能更好预测外源化合物及未识别的化合物等类别的代谢物。 3. 即该模型能识别大量未识别化合物的来源 2. 进一步的,临床数据的预测和饮食数据的预测相关性显著,而微生物组的预测与饮食数据的预测相关性最高。 3. 与遗传相关的代谢物无法被其它的组预测 4. 总结:每个特征组在一组不同的代谢物方面表现出不同的信息 5. 最后,构建了预测主要代谢物相对预测效度的模型 1. 发现饮食因素具有最强的预测效度,可推测所有特征48.9%的参数,Fig 1 e。 2. 值得注意的是,肠道菌数据具有30.8%的较高预测效度,而生活方式仅占1.9%,提示**肠道菌数据在预测及决定血清代谢物水平上具有重要意义**。 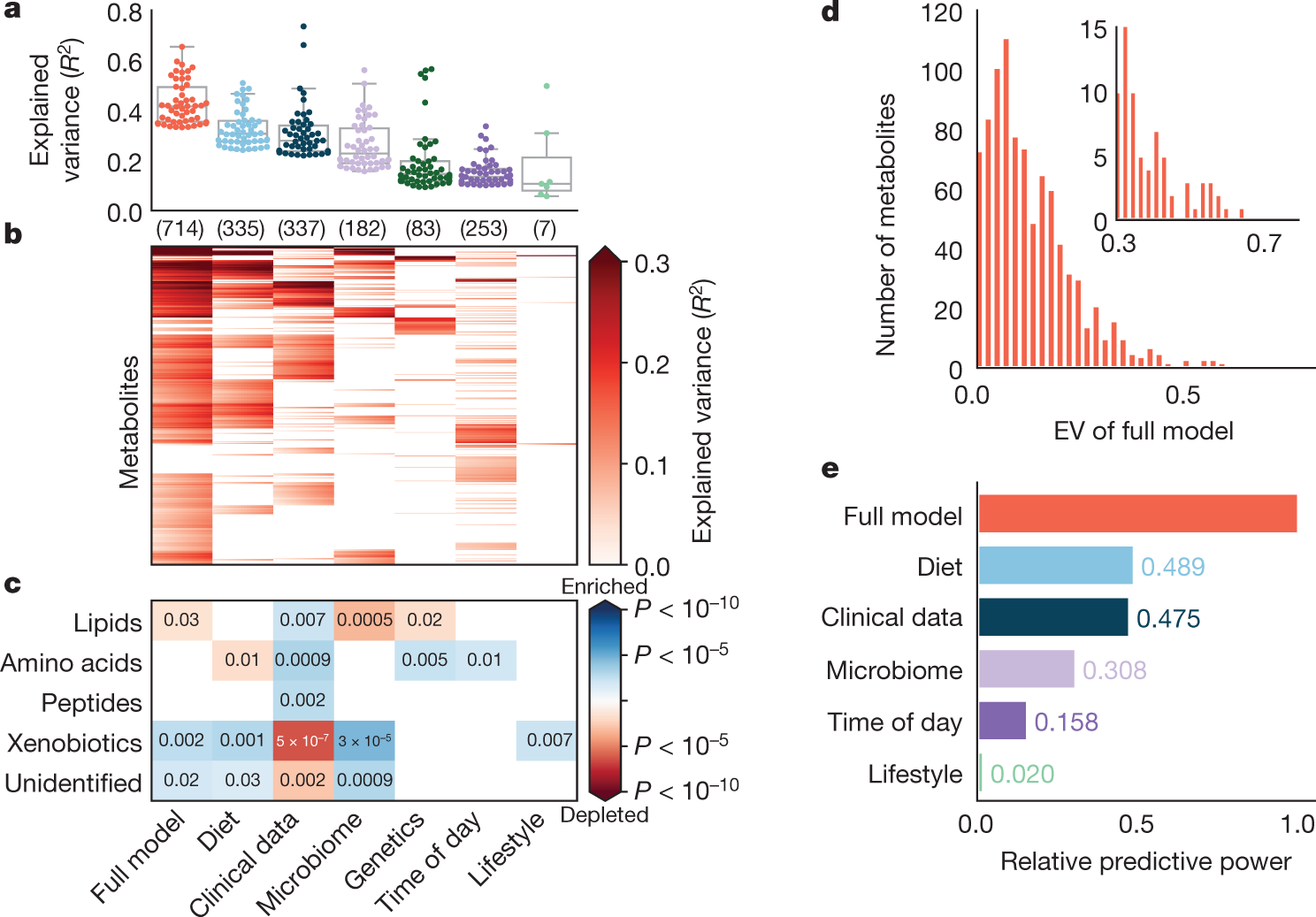 ## Replication in external cohorts,模型在外部队列的重复性 进一步验证该模型的可靠性及可重复性,研究人员使用两项地域独立的数据集对该模型的准确性进行确认。 1. 两个数据集 1. 一项来自英国TwinsUK Registry的1004名老年健康志愿者的样品 2. 一项来自IMI DIRECT cohort的北欧2型糖尿病患者的245份血清样品 2. 通过肠道菌数据特征分别获得107个或50个代谢物的预测。 3. 结果: 1. 对来自TwinsUK的107个预测代谢物中,95个可以得到验证,其中前60项均得到验证,而来自IMI DIRECT的50个预测代谢物中,28个可以得到验证 2. **本研究构建的模型可独立于人群及数据库采集方式对血清代谢物和肠道菌之间的关联进行准确预测。** 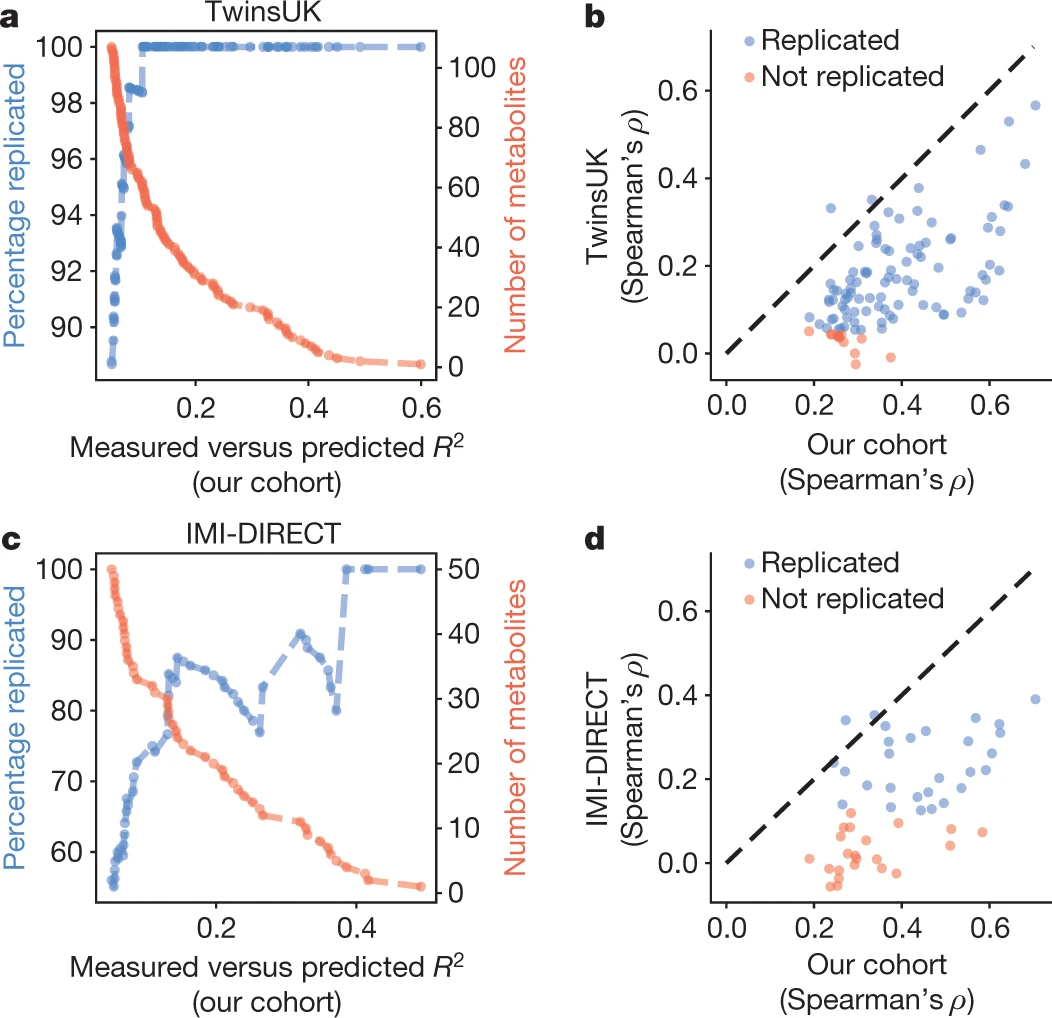 ## 饮食模型和微生物模型相互独立 1. 鉴于此前研究关于饮食调控肠道菌的结论,研究人员比较了饮食或肠道菌对血清代谢物可释方差的预测 1. 发现**除了摄取咖啡这一项可同时被饮食及肠道菌预测之外,绝大部分代谢物均可分别被饮食或肠道菌独立预测,提示饮食及肠道菌的模型具备各自独立性**,只有微生物组数据才能显著34种代谢产物 2. 即**肠道菌可独立于饮食因素来调控这些血清代谢物的生成**。 2. 为推测每个预测的驱动因素,研究人员使用基于博弈理论最优Shapley值的SHAP分析,发现多种饮食及肠道菌特征可强烈预测血清代谢物 1. 咖啡摄入可作为比其他饮食特征更强烈的预测因子来预测血清代谢物中大量外源化合物及未识别化学物的水平,这些代谢物包括黄嘌呤代谢途径的7-二甲基黄嘌呤 (paraxanthine)。 2. 另一个例子为长期摄入鱼类食物可准确预测多种血脂水平,如一种在慢性肾疾病患者血清中聚集的CMPF (一种尿毒症毒素),而这种代谢物也被报道可预防及逆转脂肪变性  3. 为评估几个重要类群是否足以正确预测,给出定义,每个代谢物的“主要预测因子”为具有最大平均绝对SHAP值的类群 1. 19个细菌类群是前50个微生物组预测代谢物的主要预测因子 2. 一种梭状芽胞杆菌是其中22种疾病的主要预测因子,这些疾病也与饮食模型中的咖啡摄入量密切相关 3. 其它分类群仅仅是一两个代谢物的主要预测因子,这**表明需要许多不同的细菌来进行准确预测** 4. 为了检验主要预测因子是否足以准确预测,将完整微生物组模型的准确性与仅基于主要预测因子的模型的准确性进行比较 1. 基于主要预测因子的模型只能解释全微生物组模型解释方差的36% 的中位数 1. 例如,肉桂酰甘氨酸,使用微生物组数据显著预测(r = 0.49,p < 10-20) ,但是,基于其主要预测因子的模型未能提供显著的预测。 2. 一些代谢物只能由一个细菌种预测 1. 例如未经确认的代谢物 X-16124,对此,以茄科物种为基础的模型解释了完整微生物组模型的93% 的方差。 2. 实际上,在检测到这种细菌的个体中,95% 的人的血清中也检测到了 X-16124,相比之下,检测不到这种细菌的个体只有23% ## 遗传-代谢组学的联系 1. 全基因组关联分析 1. 已有的研究表明,遗传学影响血清代谢 2. 研究人员也使用该模型揭示了遗传-代谢组的关联,并从 ## 概念上验证了临床干预的效度 通过将正常饮食健康志愿者进行随机分组,使其分别摄入全麦面团面包或市售白面包(Fig 5 a),研究人员随后分别在基线水平及一周干预后检测志愿者的血清代谢物,发现全麦面团面包摄入后,由本研究发现的与全麦面包正相关的标志物显著上升,平均上升1.62倍(Fig 5 c),而与全麦面包负相关的标志物平均改变仅0.66倍,而白面包干预组未检测到显著变化(Fig 5 c)。**这一实验证实了本研究构建的预测模型可有效预测不同干预措施后的血清代谢物水平改变。**  ## Discussion 尽管本文不是检测血清代谢物的大数据研究,但通过关联广泛决定因素,本文提供了一种可揭示循环血代谢物的潜在决定因素的预测模型,其中检测到的许多关联和相互作用均能有效复制此前报告的结论,证明了这个模型的有效性。 而本文也检测到了大量的新的血清代谢物的关联及相互作用,能极大地帮助我们了解健康和疾病状态下的分子机制,并帮助我们寻找改变这些血清代谢物水平的新干预措施。 1. 局限性: 1. 目前已证明药物的摄入对血液的代谢物存在影响,但测试队列由健康人组成,几乎没有药物的摄入,因此结果可能低估了药物摄入的影响 2. 除微生物组外,大多数因素的预测依然需要 replication of results 3. 缺乏可靠的注释,因此代谢物和特定的酶之间不存在关联 4. 研究基于观察性的数据,对于相互作用的解释应当谨慎,不能认为相关性等同于因果关系 ## Methods ### Description of cohorts,受试者队列 两组以色列人的库存样本,共491个。 1. 相关伦理筛查 2. 均为18-70岁之间的健康个体,进行医学、生活方式、营养方面的调查,提供粪便和血清样本进行宏基因组测序和代谢组分析,进行基因分型,进行为期一周的全面的血液检测。实时记录所有的日常活动和营养摄入。血液和粪便样本均为在严格空腹条件下采集 3. 采集时间与地点 ### Feature groups #### Diet 包括FFQ[^2]和实时记录的一周内不同食物类型的每日平均消耗量。 保留至少有1%的参与者至少消费过一次的食物。 最终为FFQ筛选出141中,实时记录筛选出670种。 #### macronutrients 每天的常量营养素,脂类、蛋白质、碳水、热量、水等的平均消耗量,通过实时记录计算 #### anthropometrics 包括体重、BMI、腰臀围和腰臀比 #### cardiometabolic 心血管代谢特征组,包括收缩压、舒张压、心率和血糖 #### drugs 包括30种二元量,以代表20中常见药物的摄入,和10个药物组(避孕药、心血管药物、抗心律失常、胃肠道药物、降脂药、非甾体抗炎药、止痛药、精神病药物、抗甲状腺药物)[^3] #### clinical data 临床数据,包括年龄、性别,以及上面的人体数据、心血管代谢特征药物特征 #### lifestyle 吸烟(现在、过去)、压力水平、实时记录的每日平均睡眠时间、锻炼时间、和午睡时间 #### time of day Boolean值,表明样本是否于当天上午采集 #### seasonal effects 取样月份,某些分析中,按季节进行分组 #### microbiome 细菌的相对丰度 ### Metabolomics profiling and preprocessing 1. 数据的获得: - 非靶液质联用,共计540份血清样本,其中19份为来自几个人的对照样本,其余521份来自于491名参与者 2. 数据的筛选 1. 剩余1170个:剔除测量次数少于10次的27种代谢物,两组队列中分布明显不同的54种代谢物 2. Log10转换,标准化(减去中位数除以标准差),去除超过5个标准差的利群样本 3. 数据的分析 1. 对于单一代谢产物,根据storage times(only for metabolites present in at least 50 samples)对代谢物水平进行回归,最后,将缺失值插补为每种代谢物的最小值 2. 按Spearman’s ρ大于0.85进行分组,得到1067个代谢物组,其中982组为单一代谢物 ### Microbiome preprocessing #### 样本采集、DNA提取、样本测序 #### 估计细菌的相对丰度 ### Comparing metabolomics to laboratory tests 1. 实验室事先测定肌酸酐和胆固醇水平,对比预测达到了代谢组学水平 2. 数据标准化 3. 对在相同抽血时采集的两个样品进行代谢组学分析和实验室测试。 ### 个体内部和之间代谢状况的相关性(微生物组) 1. 间隔一周的同一参与者的标准化代谢轮廓和不同个体的标准化代谢轮廓之间的相关性 ### 代谢物集群的预测模型(除遗传学方法之外的所有结果) 1. GBDT in LightGBM,预测分属于7个feature类的1067个代谢物群的水平 2. 以每个feature类为输入建立5倍交叉验证的EV模型,使用R2评估结果 3. 进行1000次bootstrapping迭代计算95%置信区间和p值,每次迭代执行随机的5倍交叉验证 - 每次交叉验证中,从训练集中随机替换一组相同的数据进行训练 - 使用Fisher transformation保证数据为正态 - 使用Wald test基于正态数据计算p值,并使用BH法校正FDR>10% - 评估评估模型的性能 4. 计算测量的代谢物水平和迭代得到的预测值之间的预测系数 ### 人类遗传学的预测模型 5倍交叉验证 1. 每一次验证中,计算SNP和代谢物水平之间的关联,训练模型, 1. 只对经过Bonferroni-adjusted的前10个显著的SNP进行预测 2. 若没有显著的SNP,样本取训练集的所有样本的平均值 2. 不适用bootstrapping计算95%置信区间和p值,而是估计了真实和预测代谢物水平之间Pearson相关性的P值 ### 测试SNP和代谢物之间的关系 (注释基因,对单个代谢物进行全基因组关联,适用plink计算p值和估计效力,使用conservative Bonferroni adjustment计算FDR) 1. 使用metabolion处理已命名的分子的化学鉴定、通路和子通路信息 2. 对于未知分子,使用通路分类器实现 ### 通路富集分析 Mann-whitney U检验,比较每个途径类别代谢物的预测准确性和其它没别代谢物的预测准确性 仅考虑至少存在显著预测的feature groups的metabolite groups,共有819个metabolite groups ### 基于微生物组对metabolite的预测 1. 在宏基因组测序中,分两个队列,测试了肠道微生物组和血清(circulating)代谢物之间相关性的robustness 2. 血清代谢组学是使用discovery cohort的同一代谢组学平台进行的 - 第一个validation cohort,来自TwinsUK 的1004份健康参与者样本,粪便和血液的收集间隔为0.9±1.3年 - 第二个validation cohort包括来自IMI DIRECT Association的245名欧洲血统的T2DB样本 3. # 解释 [^1]: GBDT。根据每次数据的残差,即损失函数的值。在残差减少的方向上建立一个新的模型,直到达到一定拟合精度停止。 [^2]: FFQ,food frequency questionnaire,食物频率问卷。FFQ旨在获取长期饮食习惯。 [^3]: https://doi.org/10.1038/nature25973 Loading... # 文献汇报 ## Title A reference map of potential determinants for the human serum metabolome 人血清代谢组潜在决定因素指南 https://doi.org/10.1038/s41586-020-2896-2 ## Abstract 1. 血清代谢组包含大量内源或外源的生物标志物或者疾病致病因子 1. 通常,化合物来源已知 1. 遗传性,基因,血液 2. 肠道微生物 3. 生活方式,吸烟或饮食 2. 大多数代谢物的key determinants知之甚少 2. 测量491个独特和deeply phenotyped的健康人血清样本中的1251个代谢物水平 3. ML用于预测个体的代谢物水平 1. 数据来源:遗传信息、肠道菌群、临床、饮食、生活方式和人体测量数据 2. 预测结果:超过76%的差异代谢物 3. 预测效力:饮食和肠道微生物组的预测能力最强 4. 总结:超过800代谢物的变化具有实际的意义,为理解不同条件下代谢物的变化机制和设计控制循环代谢物水平的干预措施铺平了道路 > 血清代谢组的主要研究对象是血清所含有的各种生物标志物。这些标志物既可是内源生成,也可由环境中摄取,其中部分标志物的来源较为清晰,包括高遗传性的代谢物或可被肠道菌或生活方式 (如吸烟或饮食) 所影响的代谢物,因此,血清代谢组研究对于了解多种疾病具有重要意义。 1. 首先介绍一下作者,Eran Segal,计算生物学领域的一个牛人,95-98年特拉维夫大学的理学学士学位,99-04年,斯坦福大学计算机科学和遗传学博士学位,导师Daphne Kolle,毕业后于洛克菲勒大学就职博后。博士毕业后的四年时间里,已经作为一作和通讯发表了三篇nature正刊和十几篇子刊。至今,共计近30篇NCS,子刊无数。最开始主要做转录相关方面的工作,08年左右开始做表观遗传的相关工作,随后开始进入微生物组和代谢组学的多组学结合领域。 2. 然后介绍一下文章的相关背景。血清代谢组的主要研究对象是血清所含有的各种生物标志物。这些标志物既可是内源生成,也可由环境中摄取,其中部分标志物的来源较为清晰,包括高遗传性的代谢物或可被肠道菌或生活方式 (如吸烟或饮食) 所影响的代谢物,因此,血清代谢组研究对于了解多种疾病具有重要意义。 3. 为更进一步了解血清代谢物的关键决定因素,2020年11月11日,来自以色列Weizmann科学院Eran Segal团队的研究人员在Nature杂志在线发表了题为A reference map of potential determinants for the human serum metabolome的研究论文,使用质谱技术对健康志愿者的血清样品进行了深度鉴定,基于志愿者的宿主遗传学、肠道微生物、临床参数及生活方式等特征,使用机器学习算法对血清样品中一千多种独特代谢物进行预测,从而揭示了主要代谢物的关键决定因素,从而更好地帮助我们了解这些代谢物在不同条件下变化机制,从而制定干预措施。 4. 我们看到方法概览 1.样本来源:491个健康的以色列人,18~70岁之间。 2. 数据采集:粪便、血清的宏基因组和代谢组、饮食、临床、生活方式 3.分析方式:非靶液质联用分析1251种代谢物,包括脂类、氨基酸、外源化合物、碳水化合物、肽和核苷酸,以及30%的未知化合物 ;设计预测代谢物通路的模型以对未知代谢物分类。 4.预测结果验证:对比测量数据和预测的数据,一致性高,重复性好;不同人的血清样本,并无相关性。 5.总结:本研究获得的代谢谱,提供了一种可揭示循环血代谢物的潜在决定因素的预测模型,帮助我们了解健康和疾病状态下的分子机制,并帮助我们寻找改变这些血清代谢物水平的新干预措施。 接着,分析结果 ## Main ### 方法概览 1. 临床、饮食、生活方式、遗传和肠道菌群数据事先收集 2. 非靶分析1251个代谢物水平,包括脂类、氨基酸、外源化合物、碳水化合、肽和核苷酸,以及30%的未知化合物  3. 设计可准确预测代谢物候选生物途径的模型 1. 所有样本均检测到498个代谢物,超过50%的样本中检测到1104个代谢物 2. 筛选出475名高质量数据 ## 血清代谢物的预测 > 可解释方差:很少有资料提到,以下是从代码中解读的公式,可能和实际的有些许差别。 > 可解释方差指标衡量的是所有预测值和样本之间的差的分散程度与样本本身的分散程度的相近程度。本身是分散程度的对比。最后用1-这个值,最终值越大表示预测和样本值的分散分布程度越相近。 > 1- 样本值与预测值之差的方差/样本方差  1. 血清代谢物的预测 1. 预测方法:gradient-boosted decision trees[^1],基于可释方差的系统线性模型预测志愿者数据 2. 预测结果:335个代谢物个别饮食相关特征所解释,182个代谢物可被肠道菌相关特征解释,因此**该模型对血清代谢物具有较强预测性**,Fig 1 ab。 2. 随后检测代谢物是否富集于主要预测因素superior prediction 1. 结果,Fig 1 c: 1. 临床数据能更好地预测血脂、氨基酸及多肽等类别的代谢物 2. 肠道菌数据能更好预测外源化合物及未识别的化合物等类别的代谢物。 3. 即该模型能识别大量未识别化合物的来源 2. 进一步的,临床数据的预测和饮食数据的预测相关性显著,而微生物组的预测与饮食数据的预测相关性最高。 3. 与遗传相关的代谢物无法被其它的组预测 4. 总结:每个特征组在一组不同的代谢物方面表现出不同的信息 5. 最后,构建了预测主要代谢物相对预测效度的模型 1. 发现饮食因素具有最强的预测效度,可推测所有特征48.9%的参数,Fig 1 e。 2. 值得注意的是,肠道菌数据具有30.8%的较高预测效度,而生活方式仅占1.9%,提示**肠道菌数据在预测及决定血清代谢物水平上具有重要意义**。  ## Replication in external cohorts,模型在外部队列的重复性 进一步验证该模型的可靠性及可重复性,研究人员使用两项地域独立的数据集对该模型的准确性进行确认。 1. 两个数据集 1. 一项来自英国TwinsUK Registry的1004名老年健康志愿者的样品 2. 一项来自IMI DIRECT cohort的北欧2型糖尿病患者的245份血清样品 2. 通过肠道菌数据特征分别获得107个或50个代谢物的预测。 3. 结果: 1. 对来自TwinsUK的107个预测代谢物中,95个可以得到验证,其中前60项均得到验证,而来自IMI DIRECT的50个预测代谢物中,28个可以得到验证 2. **本研究构建的模型可独立于人群及数据库采集方式对血清代谢物和肠道菌之间的关联进行准确预测。**  ## 饮食模型和微生物模型相互独立 1. 鉴于此前研究关于饮食调控肠道菌的结论,研究人员比较了饮食或肠道菌对血清代谢物可释方差的预测 1. 发现**除了摄取咖啡这一项可同时被饮食及肠道菌预测之外,绝大部分代谢物均可分别被饮食或肠道菌独立预测,提示饮食及肠道菌的模型具备各自独立性**,只有微生物组数据才能显著34种代谢产物 2. 即**肠道菌可独立于饮食因素来调控这些血清代谢物的生成**。 2. 为推测每个预测的驱动因素,研究人员使用基于博弈理论最优Shapley值的SHAP分析,发现多种饮食及肠道菌特征可强烈预测血清代谢物 1. 咖啡摄入可作为比其他饮食特征更强烈的预测因子来预测血清代谢物中大量外源化合物及未识别化学物的水平,这些代谢物包括黄嘌呤代谢途径的7-二甲基黄嘌呤 (paraxanthine)。 2. 另一个例子为长期摄入鱼类食物可准确预测多种血脂水平,如一种在慢性肾疾病患者血清中聚集的CMPF (一种尿毒症毒素),而这种代谢物也被报道可预防及逆转脂肪变性  3. 为评估几个重要类群是否足以正确预测,给出定义,每个代谢物的“主要预测因子”为具有最大平均绝对SHAP值的类群 1. 19个细菌类群是前50个微生物组预测代谢物的主要预测因子 2. 一种梭状芽胞杆菌是其中22种疾病的主要预测因子,这些疾病也与饮食模型中的咖啡摄入量密切相关 3. 其它分类群仅仅是一两个代谢物的主要预测因子,这**表明需要许多不同的细菌来进行准确预测** 4. 为了检验主要预测因子是否足以准确预测,将完整微生物组模型的准确性与仅基于主要预测因子的模型的准确性进行比较 1. 基于主要预测因子的模型只能解释全微生物组模型解释方差的36% 的中位数 1. 例如,肉桂酰甘氨酸,使用微生物组数据显著预测(r = 0.49,p < 10-20) ,但是,基于其主要预测因子的模型未能提供显著的预测。 2. 一些代谢物只能由一个细菌种预测 1. 例如未经确认的代谢物 X-16124,对此,以茄科物种为基础的模型解释了完整微生物组模型的93% 的方差。 2. 实际上,在检测到这种细菌的个体中,95% 的人的血清中也检测到了 X-16124,相比之下,检测不到这种细菌的个体只有23% ## 遗传-代谢组学的联系 1. 全基因组关联分析 1. 已有的研究表明,遗传学影响血清代谢 2. 研究人员也使用该模型揭示了遗传-代谢组的关联,并从 ## 概念上验证了临床干预的效度 通过将正常饮食健康志愿者进行随机分组,使其分别摄入全麦面团面包或市售白面包(Fig 5 a),研究人员随后分别在基线水平及一周干预后检测志愿者的血清代谢物,发现全麦面团面包摄入后,由本研究发现的与全麦面包正相关的标志物显著上升,平均上升1.62倍(Fig 5 c),而与全麦面包负相关的标志物平均改变仅0.66倍,而白面包干预组未检测到显著变化(Fig 5 c)。**这一实验证实了本研究构建的预测模型可有效预测不同干预措施后的血清代谢物水平改变。**  ## Discussion 尽管本文不是检测血清代谢物的大数据研究,但通过关联广泛决定因素,本文提供了一种可揭示循环血代谢物的潜在决定因素的预测模型,其中检测到的许多关联和相互作用均能有效复制此前报告的结论,证明了这个模型的有效性。 而本文也检测到了大量的新的血清代谢物的关联及相互作用,能极大地帮助我们了解健康和疾病状态下的分子机制,并帮助我们寻找改变这些血清代谢物水平的新干预措施。 1. 局限性: 1. 目前已证明药物的摄入对血液的代谢物存在影响,但测试队列由健康人组成,几乎没有药物的摄入,因此结果可能低估了药物摄入的影响 2. 除微生物组外,大多数因素的预测依然需要 replication of results 3. 缺乏可靠的注释,因此代谢物和特定的酶之间不存在关联 4. 研究基于观察性的数据,对于相互作用的解释应当谨慎,不能认为相关性等同于因果关系 ## Methods ### Description of cohorts,受试者队列 两组以色列人的库存样本,共491个。 1. 相关伦理筛查 2. 均为18-70岁之间的健康个体,进行医学、生活方式、营养方面的调查,提供粪便和血清样本进行宏基因组测序和代谢组分析,进行基因分型,进行为期一周的全面的血液检测。实时记录所有的日常活动和营养摄入。血液和粪便样本均为在严格空腹条件下采集 3. 采集时间与地点 ### Feature groups #### Diet 包括FFQ[^2]和实时记录的一周内不同食物类型的每日平均消耗量。 保留至少有1%的参与者至少消费过一次的食物。 最终为FFQ筛选出141中,实时记录筛选出670种。 #### macronutrients 每天的常量营养素,脂类、蛋白质、碳水、热量、水等的平均消耗量,通过实时记录计算 #### anthropometrics 包括体重、BMI、腰臀围和腰臀比 #### cardiometabolic 心血管代谢特征组,包括收缩压、舒张压、心率和血糖 #### drugs 包括30种二元量,以代表20中常见药物的摄入,和10个药物组(避孕药、心血管药物、抗心律失常、胃肠道药物、降脂药、非甾体抗炎药、止痛药、精神病药物、抗甲状腺药物)[^3] #### clinical data 临床数据,包括年龄、性别,以及上面的人体数据、心血管代谢特征药物特征 #### lifestyle 吸烟(现在、过去)、压力水平、实时记录的每日平均睡眠时间、锻炼时间、和午睡时间 #### time of day Boolean值,表明样本是否于当天上午采集 #### seasonal effects 取样月份,某些分析中,按季节进行分组 #### microbiome 细菌的相对丰度 ### Metabolomics profiling and preprocessing 1. 数据的获得: - 非靶液质联用,共计540份血清样本,其中19份为来自几个人的对照样本,其余521份来自于491名参与者 2. 数据的筛选 1. 剩余1170个:剔除测量次数少于10次的27种代谢物,两组队列中分布明显不同的54种代谢物 2. Log10转换,标准化(减去中位数除以标准差),去除超过5个标准差的利群样本 3. 数据的分析 1. 对于单一代谢产物,根据storage times(only for metabolites present in at least 50 samples)对代谢物水平进行回归,最后,将缺失值插补为每种代谢物的最小值 2. 按Spearman’s ρ大于0.85进行分组,得到1067个代谢物组,其中982组为单一代谢物 ### Microbiome preprocessing #### 样本采集、DNA提取、样本测序 #### 估计细菌的相对丰度 ### Comparing metabolomics to laboratory tests 1. 实验室事先测定肌酸酐和胆固醇水平,对比预测达到了代谢组学水平 2. 数据标准化 3. 对在相同抽血时采集的两个样品进行代谢组学分析和实验室测试。 ### 个体内部和之间代谢状况的相关性(微生物组) 1. 间隔一周的同一参与者的标准化代谢轮廓和不同个体的标准化代谢轮廓之间的相关性 ### 代谢物集群的预测模型(除遗传学方法之外的所有结果) 1. GBDT in LightGBM,预测分属于7个feature类的1067个代谢物群的水平 2. 以每个feature类为输入建立5倍交叉验证的EV模型,使用R2评估结果 3. 进行1000次bootstrapping迭代计算95%置信区间和p值,每次迭代执行随机的5倍交叉验证 - 每次交叉验证中,从训练集中随机替换一组相同的数据进行训练 - 使用Fisher transformation保证数据为正态 - 使用Wald test基于正态数据计算p值,并使用BH法校正FDR>10% - 评估评估模型的性能 4. 计算测量的代谢物水平和迭代得到的预测值之间的预测系数 ### 人类遗传学的预测模型 5倍交叉验证 1. 每一次验证中,计算SNP和代谢物水平之间的关联,训练模型, 1. 只对经过Bonferroni-adjusted的前10个显著的SNP进行预测 2. 若没有显著的SNP,样本取训练集的所有样本的平均值 2. 不适用bootstrapping计算95%置信区间和p值,而是估计了真实和预测代谢物水平之间Pearson相关性的P值 ### 测试SNP和代谢物之间的关系 (注释基因,对单个代谢物进行全基因组关联,适用plink计算p值和估计效力,使用conservative Bonferroni adjustment计算FDR) 1. 使用metabolion处理已命名的分子的化学鉴定、通路和子通路信息 2. 对于未知分子,使用通路分类器实现 ### 通路富集分析 Mann-whitney U检验,比较每个途径类别代谢物的预测准确性和其它没别代谢物的预测准确性 仅考虑至少存在显著预测的feature groups的metabolite groups,共有819个metabolite groups ### 基于微生物组对metabolite的预测 1. 在宏基因组测序中,分两个队列,测试了肠道微生物组和血清(circulating)代谢物之间相关性的robustness 2. 血清代谢组学是使用discovery cohort的同一代谢组学平台进行的 - 第一个validation cohort,来自TwinsUK 的1004份健康参与者样本,粪便和血液的收集间隔为0.9±1.3年 - 第二个validation cohort包括来自IMI DIRECT Association的245名欧洲血统的T2DB样本 3. # 解释 [^1]: GBDT。根据每次数据的残差,即损失函数的值。在残差减少的方向上建立一个新的模型,直到达到一定拟合精度停止。 [^2]: FFQ,food frequency questionnaire,食物频率问卷。FFQ旨在获取长期饮食习惯。 [^3]: https://doi.org/10.1038/nature25973 最后修改:2022 年 10 月 02 日 © 允许规范转载 赞 如果觉得我的文章对你有用,请随意赞赏